Summary
Epithelioid sarcoma (ES) is an aggressive mesenchymal neoplasm with a poor prognosis. Based on clinical and histological features, the tumor is divided into proximal and distal subtypes. Both subtypes are characterized by the loss of SMARCB1 expression. The tumor is frequently misdiagnosed. The principal distal sites are the fingers, hands, and forearms. Prompt diagnosis followed by aggressive surgical resection with wide or radical margins is the most effective treatment.
Complete Information on this Tumor
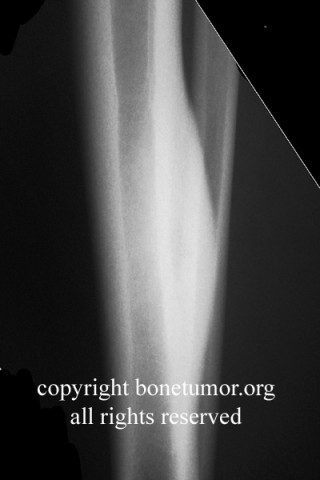
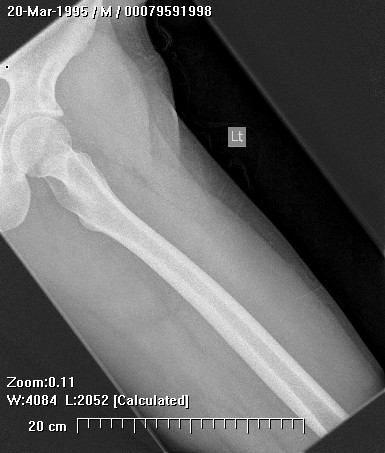
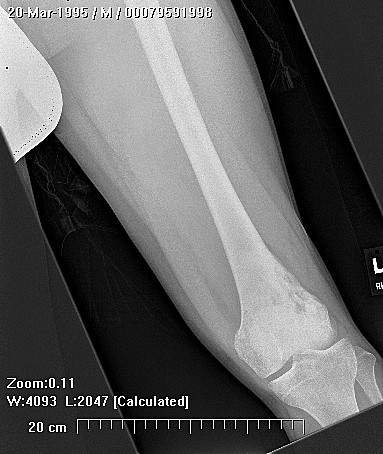
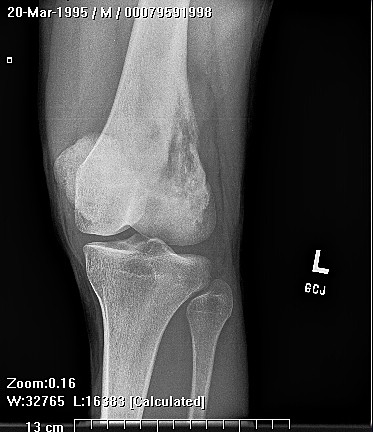
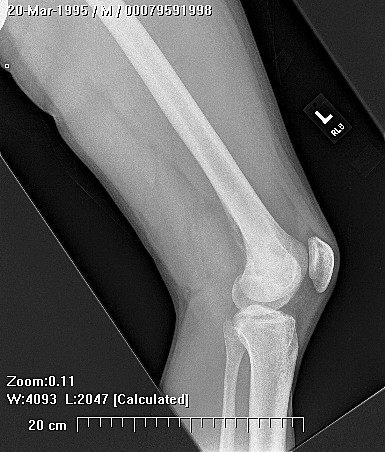
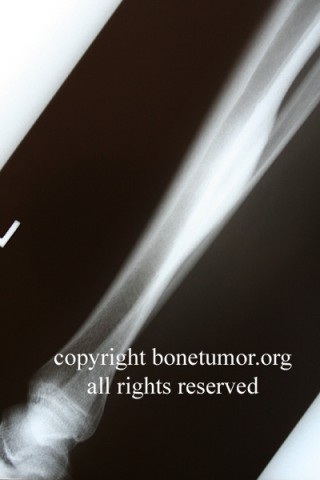
Epithelioid sarcoma (ES) is an aggressive mesenchymal neoplasm with a poor prognosis. Based on clinical and histological features, the tumor is divided into proximal and distal subtypes. The benign, cutaneous clinical presentation leads to a high risk of misdiagnosis and delay. Although this sarcoma is generally rare, the most common location for the distal subtype is the distal upper and lower extremities, with approximately 15% of all cases located in the distal lower extremity. The tumor grows and spreads along lymph and vascular channels as well as tendon sheaths leading to more generalized swelling and a permeative mass. The most common sites are the fingers, hands, and forearms. The tumor spreads to lymph nodes, proximal soft tissue sites, and to the lung. Epithelioid sarcoma is the most common soft tissue sarcoma in the hand and wrist, followed by alveolar rhabdomyosarcoma and synovial sarcoma.
The tumor may present as a small, firm superficial or deep nodule or a focal cluster of nodules. Regional multifocal presentation is an unusual characteristic displayed by this tumor. This tumor is frequently misdiagnosed as a skin condition, warts, or corns, and a correct diagnosis may be delayed with serious medical and legal consequences. About one half of the tumors are not painful. The tumor occurs in both subcutis and deeper tissues. When located in the subcutis, it usually presents as a firm nodule that may be solitary or multiple, has a calluslike consistency, and is often described as a “woody hard knot” or :firm lump” that is slow growing and painless. Nodules situated in the dermis are often elevated above the skin surface and frequently become ulcerated weeks or months after they are first noted. Such lesions are often erroneously diagnosed as an “indurated ulcer”, “draining abscess”, or “infected wart” that fails to heal despite intensive therapy. The majority of tumors are 3 to 6 cm in diameter.
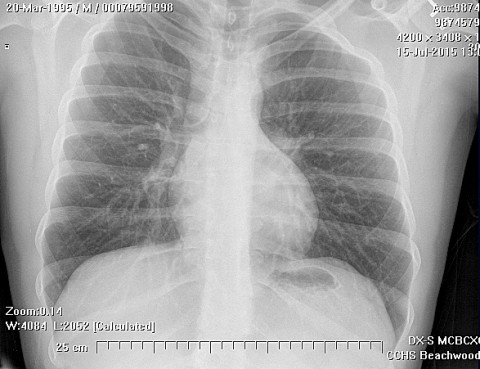
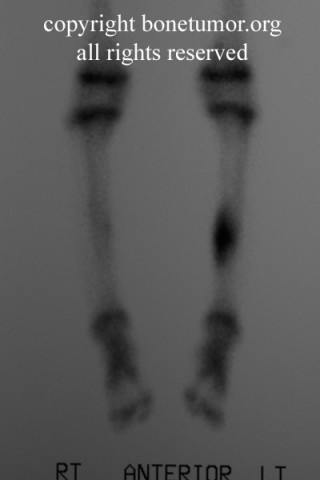
Lymph node metastasis should be evaluated at the time of presentation, due to the unusual propensity of this tumor to spread to regional nodes. In general, lymph node metastasis in sarcomas are quite rare, occurring in about 2.6% of all sarcoma patients in large series. In ES, however, the rate of lymph node metastasis is around 30% (range 14 - 44%) Lung metastasis occurs in 20-40% of patients.
The principal microscopic characteristics are the distinct nodular arrangement of the tumor cells, their tendency to undergo central degeneration and necrosis, and their epithelioid appearance and eosinophilia. The nodular pattern, probably the most conspicuous single feature of epithelioid sarcoma, varies somewhat; in some tumors the nodules are well circumscribed; in others they are less well defined and are often compacted into irregular multinodular masses. Necrosis of the tumor nodules is a common finding; it is most prominent in the center of the nodules and at times is associated with hemorrhage and cystic change. When tumor spreads within a fascia or aponeurosis, it forms festoonlike or garlandlike bands punctuated by areas of necrosis.
The constituent cellular elements range from large ovoid or polygonal cells with deply eosinophilic cytoplasm, suggesting a rhabdomyosarcoma or malignant rhabdoid tumor, to plump spindle-shaped cells reminiscent of fibrosarcoma or malignant fibrous histiocytoma. In some of the latter tumors the spindle cell pattern may predominate and may obscure the characteristic epithelioid features and nodularity.
Cytogenetic analysis of an epithelioid sarcoma cell line revealed a karyotype of 64 to 66 chromosomes with extensive numerical and structural rearrangements and up to 24 marker chromosomes.
Immunohistochemical findings
The cells show coexpression for low-molecular-weight (45 kd and 54 kd) and high- molecular- weight (57 kd) cytokeratin, vimentin, and epithelial membrane antigen.
Ultrastructural findings
Most investigators report polygonal and spindle shaped cells with ovoid, indented nuclei having small amounts of marginally places chromatin. The cytoplasm contains arrays of rough endplasmic reticulum, a prominent Golgi apparatus, and free ribosomes as well as occasional mitochondria, lysosomes, and droplets of osmophilic material. Intermediate filaments are a common and striking feature. They may be arranged longitudinally as in myofibroblasts or more often from paranuclear masses or whorls, a feature that probably accounts for the voluminous cytoplasm and the striking epithelioid appearance and eosinophilia of the tumor cells.
Surgical treatment requires early radical excision or amputation if the primary tumor is situated in the fingers or toes. Treatment options for tumors which have metastasized are much more limited. Limb salvage may be possible in selected cases.
For surgically controllable disease, surgical removal with a very wide or radical margin is mandatory. Local and regional metastasis may necessitate amputation. hand and palm lesions are best treated with complete amputation. Amputation should also be considered as treatment for recurrent growth, but does not seem to offer any benefit to patients with distant metastasis.
In all cases surgical treatment should be combined with radiotherapy and multiagent chemotherapy over a prolonged period.
Second, the lesion affects the hand and wrist in a large proportion of cases, and the initial surgical management may be performed by a hand specialist. Hand and wrist surgeons may feel obligated to preserve sensation and function, rather than to radically resect tumors and nearby structures that may be essential, such as bone, nerve, and tendon. This may lead to inadequate initial surgical margins. Inadequate surgical margins are known to increases the risk of local recurrence and tumor metastasis. In general, many surgeons are reluctant to perform the radical procedures required to control this tumor, and local recurrence and regional or systemic metastasis may occur.
The potential of the tumor to recur and metastasize is clearly evident. Most common sites of sites are lymph nodes and the skin, and the scalp. Multiple recurrences, often a result of marginal resection are a characteristic feature of the tumor. The recurrent tumor generally presents as confluent nodules in the dermis or along tendons and fascial structures at or near the original tumor site. Recurrence generally develops within first year after diagnosis, but may be late. Metastases may manifest before detection of the primary tumor.