Summary
This tumor has been given the names benign fibrous hystiocytoma, fibrous histiocytoma, xanthofibroma, fibroxanthoma of bone, and primary xanthoma of bone. The author of this site prefes the name benign fibrous histiocytoma.
Complete Information on this Tumor
This tumor has been given the names benign fibrous hystiocytoma, fibrous histiocytoma, xanthofibroma, fibroxanthoma of bone, and primary xanthoma of bone. The author of this site prefes the name benign fibrous histiocytoma.
There is disagreement amongst pathologists as to whether this tumor represents a true neoplasm, adevelopmental defect, or a reactive process. A portion of the confusion arises from the lack of agreement between pathologists as to what exactly defines this lesion. Different pathological, clinical and radiographic definitions of this lesion have led to different findings of frequency, age range, location in the skeleton, histological appearance, and tumor behavior. The authors of this site have combined information from their own personal experience and a number of published sources to create this summary. For additional information please refer to the primary sources listed in the references section.
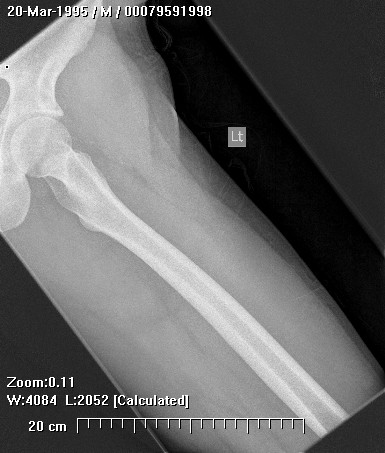
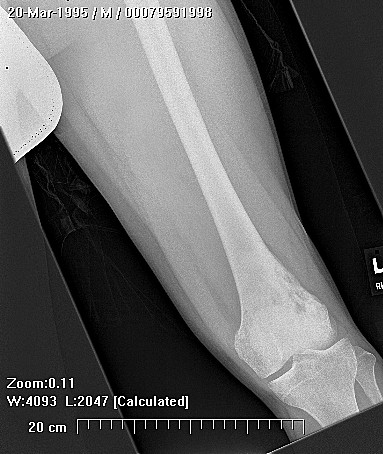
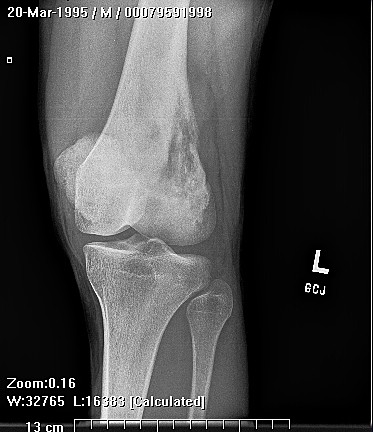
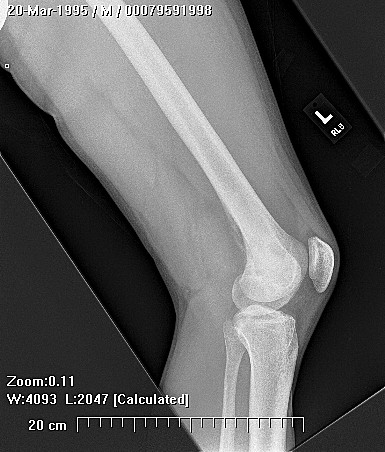
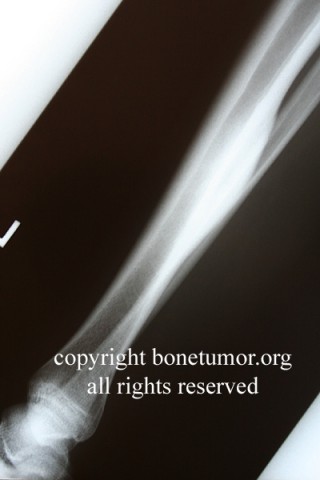
Clinically, patients report pain from the lesion, often of months or years duration. Pain may be associated with pathological fracture. There may be some local tenderness, but no swelling or mass is seen, and there are no systemic symptoms. There is normally no impairment of the function of the nearby joint. Spinal lesions may cause neurologic defect by pressing on the spinal cord. In some cases there is a primary underlying disorder of cholesterol metabolism or other lipid abnormalities. In these cases the lytic bone lesions are analogous to those seen in storage diseases such as Gaucher's disease. These multiple lesions are termed "xanthoma disseminatum". One reported case is of a 10 year old boy with lytic lesions in the pelvis, femur, and humerus, as well as yellow and brown papules and plaques on the face and trunk. This patient also had polyuria and polydipsia, and was found to have diabetes insipidus. (Khandpur) Radiographically, the lesion occur commonly in the ribs, pelvis, including the sacrum and ilium, or in the epiphysis or diaphysis of tubular bones. These tumors have been reported in the jaw and associated soft tissues. In another report this tumor occurred commonly around the knee.
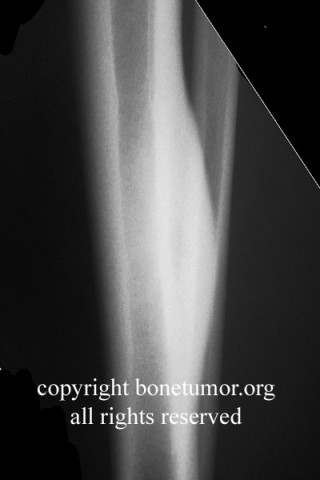
CT scan shows a moderately irregular lytic area with an prominent trabecular pattern and surrounding sclerotic bone.
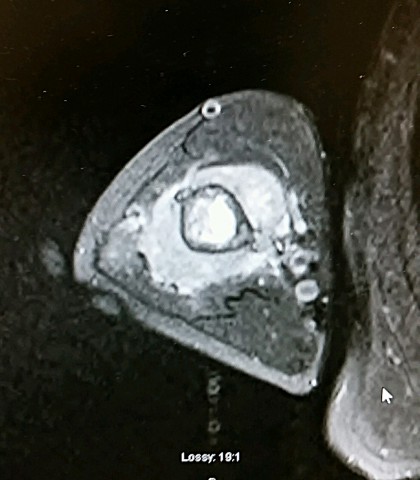
On MRI scans, there is central low signal intensity with a surrounding rim of high signal on T1, and more uniform but somewhat varigated high signal intensity on T2 sequences, with the surrounding sclerotic bone having low signal intensity.
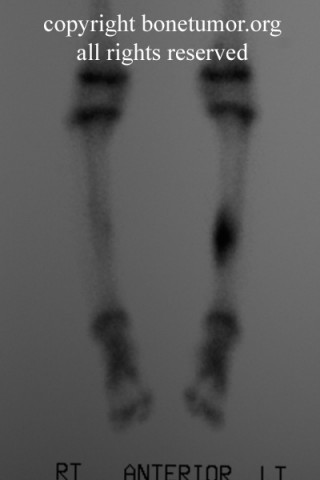
On bone scan, the lesion has been reported to have no uptake, but the author cannot confirm this.
Clarke et al, Am J Surg Pathol., 1985;9:806-815
Macdonald, Arch Pathol Lab Med. 2002;126:599-601
Khandpur et al, Aus J Dermatol., 2003;44:190