Summary
Description
Ewing's sarcoma is a highly malignant tumor that affects mostly children. It is most commonly found femur, tibia and humerus, as well as the pelvis.
People and Age
People ages 1 to 20 are most commonly affected by this disease; however, a broad range of ages may be affected.
Symptoms and Presentation
The clinical presentation of Ewing's sarcoma includes pain and swelling of weeks or months duration.
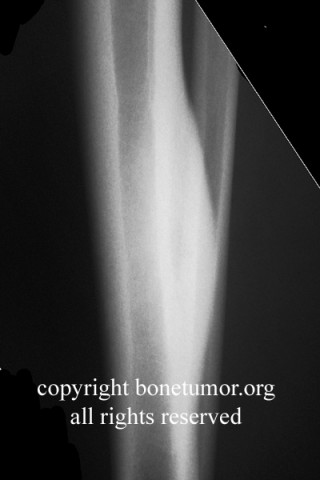
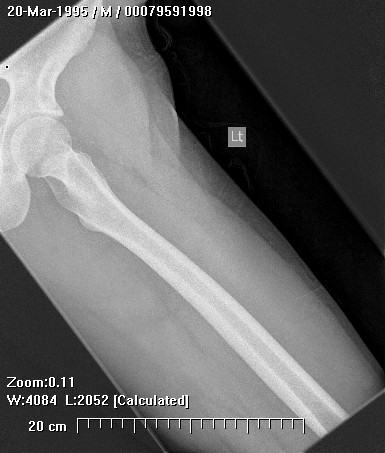
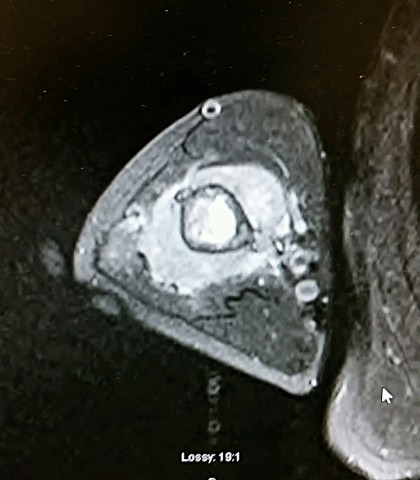
Brief description of the xray
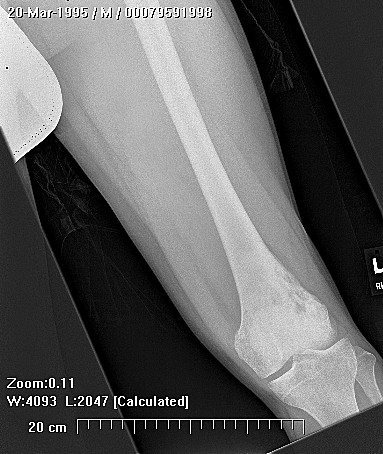
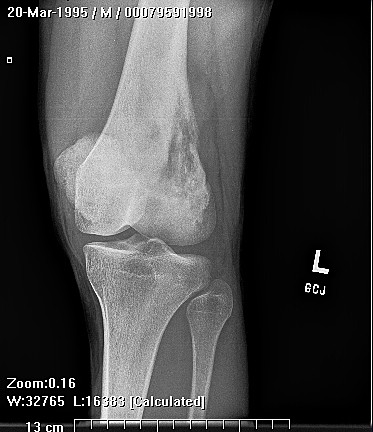
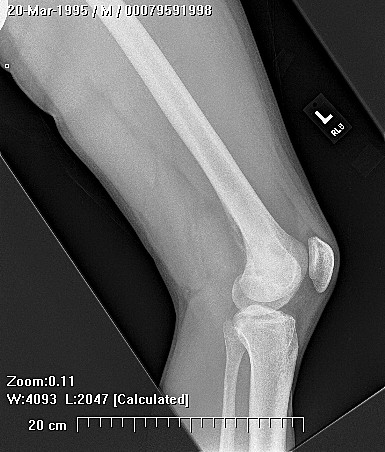
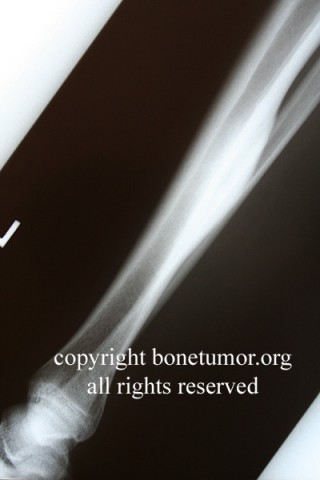
X-rays show the appearrance of permeative margins due to presence of the tumor in the Haversian Canals. Ewing's Sarcoma is not always obvious on X-ray and other diagnostic methods may be needed for diagnosis.
Complete Information on this Tumor
Introduction and Definition
Ewing's sarcoma is a highly malignant tumor that is a type of peripheral primitive neuroectodermal tumor (PNET). Ewing's sarcoma is found in the lower extremity more than the upper extremity, but any long tubular bone may be affected. The most common sites are the metaphysis and diaphysis of the femur followed by the tibia and humerus.
Incidence and Demographics
Ewing's sarcoma is most common in the first and second decade but may affect persons from age 2 to 80. This tumor preferentially affects whites more than blacks and Asians. The ratio of male to female is 3:2.
Symptoms and Presentation
The clinical presentation of Ewing's sarcoma includes pain and swelling of weeks or months duration. Erythema and warmth of the local area are sometimes seen. Osteomyelitis is often the initial diagnosis based on intermittent fevers, leukocytosis, anemia and an increased ESR.
X-Ray Appearance and Advanced Imaging Findings
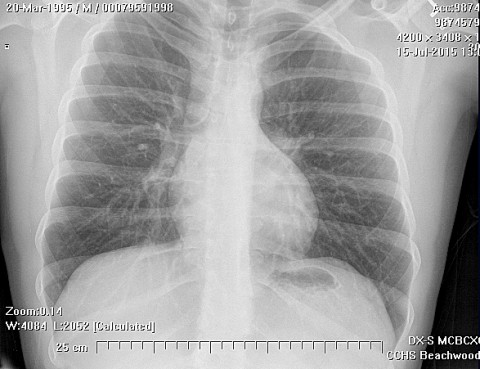
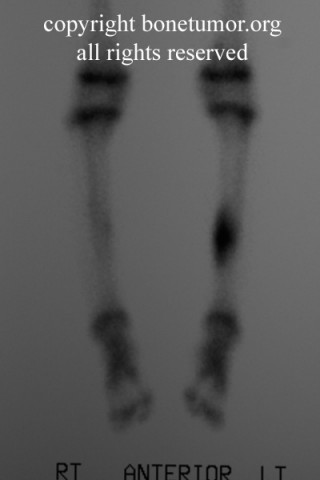
Radiologically, Ewing's sarcoma is often associated with a lamellated or "onion skin" periosteal reaction. This appearance is caused by and splitting and thickening of the cortex by tumor cells. The lesion is usually lytic and central. Endosteal scalloping is often present. The "onion-skin" appearance is often followed with a "moth-eaten" or mottled appearance and extension into soft tissue. Bone marrow infiltration is not obvious on plain x-ray. While Ewing's sarcoma is usually lytic it may present as a sclerotic lesion with bone expansion. CT is helpful in defining bone destruction. MRI is essential to elucidate the soft tissue involvement; TI-weighted images the tumor has low intensity compared to the normal high intensity of bone marrow. On 1:2 -weighted images the tumor is hyper intense compared to muscle. Ewing's sarcoma has increased uptake on bone scan.
Differential Diagnosis
Infection, neuroblastoma metastasis, lymphoma, leukemia
Preferred Biopsy Technique for this Tumor
Open biopsy for bone lesions, core or tru-cut is often adequate for soft tissue lesions, if the pathologist has significant experience with sarcomas.
Histopathology findings
Grossly, the tumor is gray to white in color and poorly demarcated. The consistency is soft and gray and sometimes semi-liquid especially after breaking through the cortex. Areas of hemorrhage and necrosis are common. The destruction is often greater on gross appearance than was visible on radiographs.Under the microscope, Ewing's sarcoma consists of densely packed uniform small cells in sheets. The cells have scant cytoplasm without distinct borders. The cells are two to three times as big as lymphocytes and have a single oval or round nucleus without prominent nucleoli. The tumor spreads through Haversian canals which cause the appearance of permeative margins on x-ray. Glycogen is present within the cells causing a positive reaction to periodic acid-schiff (PAS) stain. Most Ewing's sarcomas are positive with HBA-71 or 0-13 stain which is an antibody to the protein product of myc 2. The microscopic differential includes lymphoma and metastatic neuroblastoma which must be excluded by reticulin stain and urine vanillyl mandelic acid and homovanillic acid respectively. Rhabdomyosarcoma is ruled out if the specimen stains negatively with desmin, myoglobin and actin stains. A neural origin is supported by electron microscope findings of pseudorosettes. This is further supported by the common finding in Ewing's sarcoma and primitive neuroectodermal tumors of choline acetyltransferase and the translocation t(11:22)(q24;ql2). It is thought that Ewing's sarcoma with its few organelles is the poorly differentiated end of the spectrum of PNET. Neuroepithelioma is an example of well differentiated PNET and has neurosecretory granules and neuritic processes.
Treatment Options for this Tumor
Treatment for Ewing's sarcoma includes surgery, radiation and multi-drug chemotherapy. Radiation or chemotherapy with vincristine, dactinomycin and cyclophophamide (VAC) are used preoperatively. Adjuvant chemotherapy follows surgery and decreases recurrences. The tumor can metastasize to lungs and lymph nodes. Poor prognostic signs include increased age, increased ESR and leukocytosis at presentation.
Preferred Margin for this Tumor
Wide
Special and Unusual Features
Poor prognostic signs include increased age, increased ESR and leukocytosis at presentation.
Suggested Reading and Reference
Eggli, KD et al., Ewing's Sarcoma, Radiologic Clinics of North America, 31(2):325-337, March, 1994.
Bulloughs, Peter, Orthopaedic Pathologv (third edition), Times Mirror International Publishers Limited, London, 1997.
Fletcher, Christopher, Diagnostic Histopathology of Tumors, Churchill Livingstone:New York, 1990.
Huvos, Andrew, Bone Tumors:Diagnosis Treatment and Prognosis, W.B.Saunders, Co., 1991.