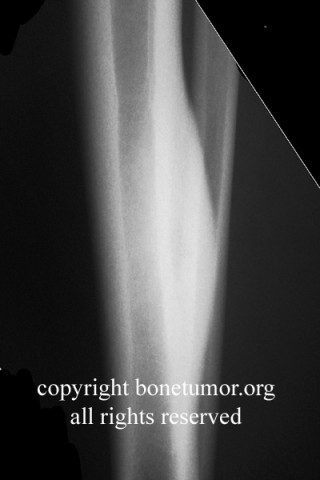
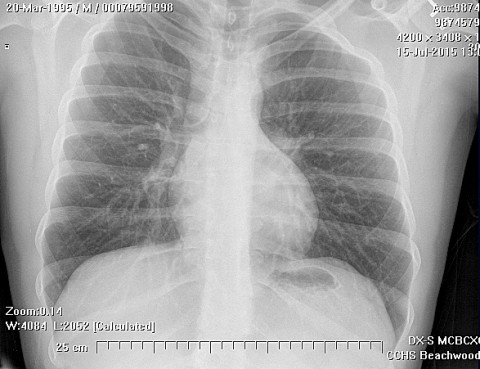
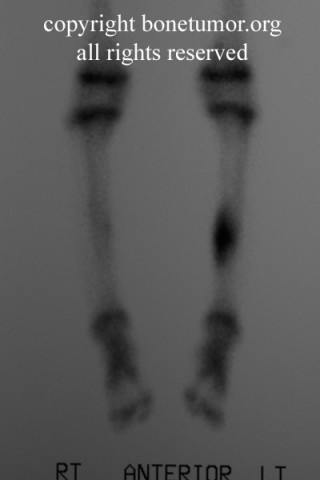
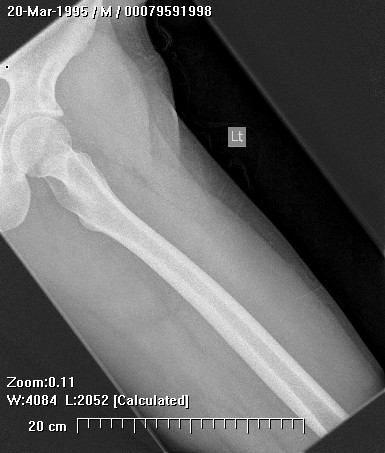
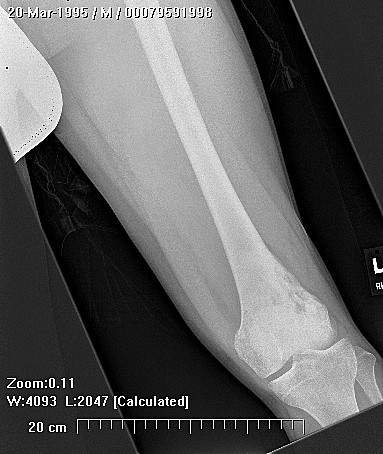
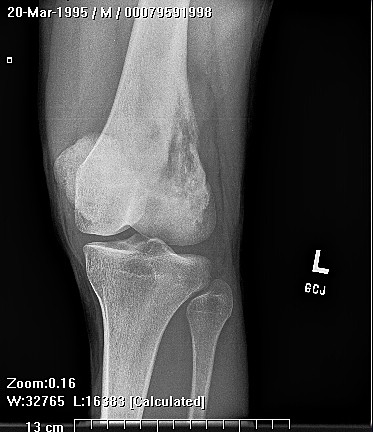
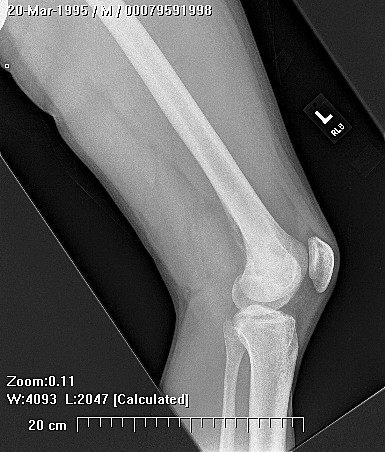
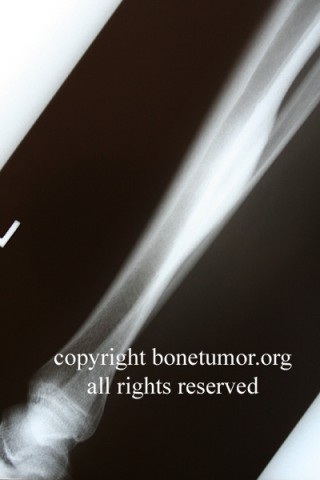
Chondrosarcoma is a rare disease, with an estimated incidence of 1 in 200000 per year. There is inconsistency in the literature with regard to the factors affecting the survival rate because many reports have focused on relatively small single institutional experiences. Although tumor grade and surgical stage are assumed to be the most significant prognostic variables in modern clinical practice, there has been disagreement regarding this assumption.
Chondrosarcoma has been deemed a surgical disease for many decades, and surgery remained the primary treatment modality over the entire data collection period. Chemotherapy and radiation have not been routinely utilized for the treatment of this disease.
The Surveillance, Epidemiology and End Results (SEER) Program of the National Cancer Institute represents retrospective data on a total of 2890 patients. Data was collected from 1973 to 2000 years.
The mean age at the time of diagnosis was fifty-one years (range one to 102 years). The sex distribution demonstrates slight male predominance (55%). The tumor location was most commonly classified as appendicular (44.5%), followed by axial (31.1%), and then by soft tissue (9.9%).
Low grade chondrosarcomas, comprising Grade-1 tumors (901 patients; 31.5%) and Grade-2 tumors (926 patients; 32.0%), were more frequent than high-grade chondrosarcomas, comprising Grade-3 tumors (207 patients; 7.2%) and Grade-4 tumors (163 patients; 5.7%0.
In the first four months after the diagnosis, 2468 patients (85.4%) were manged operatively and 373 patients (12.9%) were manged nonoperatively. Radiation therapy was received by 346 (12%) of the patients.
Most tumors (2387 patients; 82.6%) were classified as chondrosarcoma NOS (not otherwise specified). Other reported subtypes, in order of increasing frequency, where myxoid (283 patients; 9.8%), mesenchymal (126 patients; 4.4%), dedifferentiated (40 patients; 1.4%), juxtacortical (21 patients; 0.7%), and clear cell (13 patients; 0.4%) variants. Malignant chondroblastoma (20 patients; 0.7%) was also included in the study. The five year survival rates according to histological type varied widely, from 0% (dedifferentiated subtype) to 100% (clear cell subtype). In the group of patients with dedifferentiated subtype, there were no survivors after 3 years. Patients with chondrosarcoma NOS had a five year survival rate of 70% similar to the rate for those with the myxoid subtype (71%). Juxtacortical chondrosarcoma and malignant chondroblastoma were associated with slightly better survival rates (93% and 85% respectively), and mesenchymal chondrosarcoma was associated with a slightly worse survival rate (48%). Subtypes with a worse survival rate were associated with a higher percentage of hi-grade tumors (85.3% for dedifferentiated chondrosarcoma and 77.8% for mesenchymal chondrosarcoma).
The disease-specific and overall survival rate stabilized at about 10 years after the diagnosis and remained at 72.8% at 30 years. The overall survival rate, however, continued to decline steadily after diagnosis and reached 33.3% at 30 years. This data indicate that patients who survived the first 10 years after diagnosis are more likely to die of other causes than they are to die from chondrosarcoma-related events.
With regard to sex, women had a slightly better survival rate than men did at 30 years of follow up (78.6% compared with 68.7%).
The rate of survival at 30 years was 76% for patients with low-grade tumors, compared with 50% for those with high-grade tumors. In the group of patients with high-grade tumors, there was an accelerated mortality rate in the first 10 years, after which their survival rate stabilized. The group of patients with low-grade tumors had a more gradual attenuation in the survival rate over time.
Patients were grouped according to whether the disease status was localized, regional, or distant, which reflect the M0, M1, and M2 classifications of the AJCC staging system. Patients with localized disease (M0) had twice the 30-year survival rate of those with regional disease (M1) (43% compared with 22.3%). The latter patients had twice the 30-year survival rate of patients with metastatic disease (M2) (22.3% compared with < 10%).
Survival rate stratified according to age at the time of diagnosis: patients who were 50 years old or less had a significantly better disease-free survival rate as well as a significantly better overall survival rate (> 60%) after 30 years than did patients who were more than 50 years old (< 20%).
When the significant univariate variables of histological grade, surgical stage, sex, and tumor site were evaluated, only histological grade and surgical stage were shown to be independent predictors of survival.
The relative 5 year survival rates for conventional, myxoid, juxtacortical, and mesenchymal chondrosarcoma were 70%, 71%, 87%, and 52% respectively.
The limitations of the present study are related to the SEER database and include the lack of information on how the cancer was detected, patient comorbidities, and treatment provided more than 4 months after the diagnosis. After surviving 10 years of this disease, it is unlikely that chondrosarcoma will be the ultimate cause of the patient's death.
Topic Presentation